Services
|
Protein identification (in-gel)
The workflow includes in-gel digestion, peptide purification via StageTips (Rappsilber et al, Nat. Prot. 2007), LC-MS/MS analysis (short gradient, 45 min) and database search (Sequest search engine, Proteome Discoverer 2.1). Expected results: protein identification from any protein detectable by silver or Coomassie staining. Output: List of identified proteins (min 2 peptides at 99% confidence), mgf file on request. |
|
Protein identification (in-solution)
If possible, in-solution protein digestion is the most straightforward way in order to identify purified proteins. The workflow includes in-solution digestion, peptide purification via StageTips (Rappsilber et al, Nat. Prot. 2007), LC-MS/MS analysis (short gradient, 45 min) and database search (Sequest search engine, Proteome Discoverer 2.1). Expected results: protein identification from as low as 1 ng of starting purified protein. Output: List of identified proteins (min 2 peptides at 99% confidence), mgf file on request. |
|
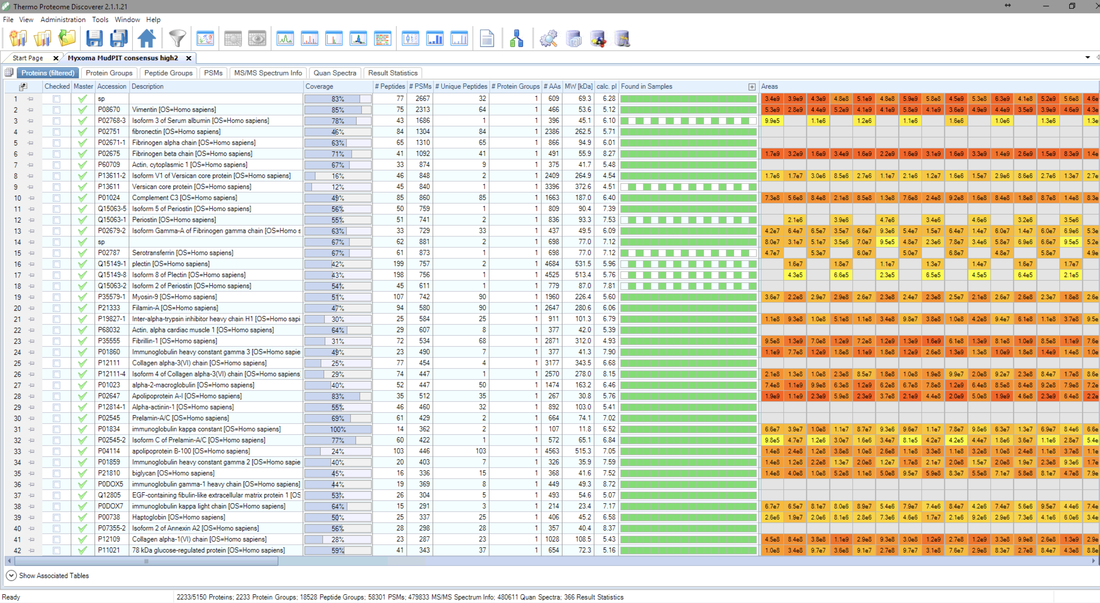
Bottom-up proteomics (qualitative)
Though quantitative analysis is generally the ultimate purpose of proteomics, in some cases a simple list of detectable proteins in a sample may be informative. The workflow includes protein precipitation, in-solution digestion, peptide purification via StageTips (Rappsilber et al, Nat. Prot. 2007), LC-MS/MS analysis (long gradient, 90 min) and database search (Sequest search engine, Proteome Discoverer 2.1). Expected results: Identification of 1500-2500 proteins (cell lysate) from a minimum of 1 microgram of total proteins. Output: List of identified proteins (min 2 peptides at 99% confidence, protein FDR better than 1%), mgf file on request. |
|
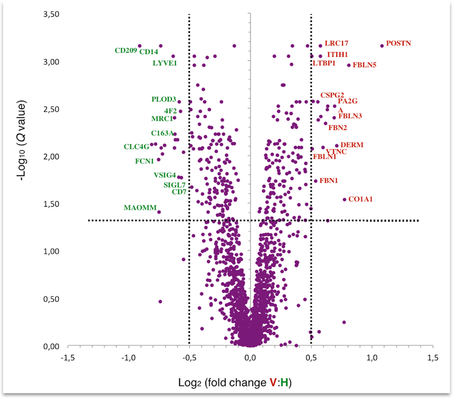
Quantitative proteomic analysis
Quantitative proteomics by mass spectrometry can rely on isotopic labeling (TMT, dimethyl labeling, SILAC) or on label-free analysis. Our proposed workflow consists in: (i) protein digestion; (ii) nLC-MS/MS analysis with long gradients (>120 min); (iii) database search and label-free quantification (Maxquant software). The implementation of alternative approaches (TMT, dimethyl labeling, SILAC) is feasible (please enquire for prices). Expected results: quantification of at least 3000 proteins based on at least one unique peptide (protein FDR better than 1%). Output: Protein identification and quantification tables; peptide data. |
|
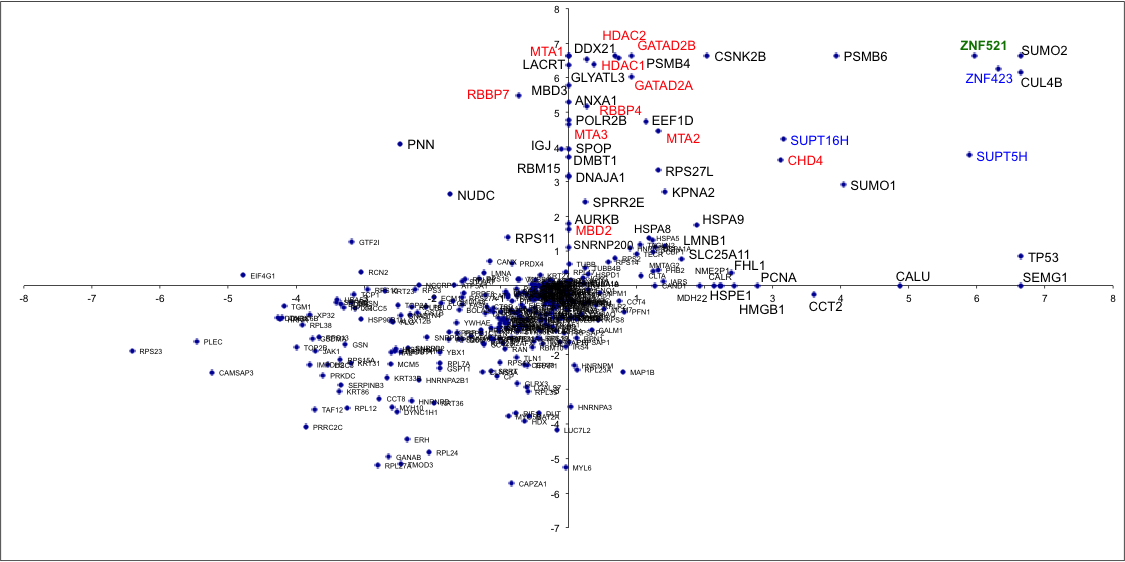
Protein interaction analysis
Affinity enrichment- Mass spectrometry (AE-MS) has become an essential tool for elucidating protein-protein interactions. We can provide quantitative comparison between the immunoprecipitate of your protein of interest and an appropriately chosen control. The workflow relies on on-bead digestion and nLC-MS/MS analysis in label free mode, and it is generally applicable to almost any type of enrichment strategy. Expected results: identification of 1000 proteins (mostly a-specific background); detection of (typically) 10-50 enriched proteins. Output: Protein identification and quantification tables; peptide data. |